Repurposing Protein Folding Models for Generation with Latent Diffusion
Quick Answer
PLAID is a multimodal generative model that generates protein sequences and structures by sampling from the latent space of protein folding models.
Quick Take
PLAID is a multimodal generative model that generates protein sequences and structures by sampling from the latent space of protein folding models. It addresses challenges in all-atom generation and organism specificity, utilizing sequence databases that are significantly larger than structural ones. This approach aims to streamline drug discovery by controlling protein generation through a textual interface.
Key Points
- PLAID generates both protein sequences and 3D structures simultaneously.
- It utilizes sequence databases 2-4 orders of magnitude larger than structure databases.
- The model addresses multimodal generation challenges in drug design.
- Training requires only sequence data, making it cost-effective and efficient.
- PLAID mirrors image generation control through a textual interface for proteins.
Paper Resources
Article Content
From source RSS / original summaryPLAID is a multimodal generative model that simultaneously generates protein 1D sequence and 3D structure, by learning the latent space of protein folding models. The awarding of the 2024 Nobel Prize to AlphaFold2 marks an important moment of recognition for the of AI role in biology. What comes next after protein folding? In PLAID, we develop a method that learns to sample from the latent space of protein folding models to generate new proteins.
It can accept compositional function and organism prompts, and can be trained on sequence databases, which are 2-4 orders of magnitude larger than structure databases. Unlike many previous protein structure generative models, PLAID addresses the multimodal co-generation problem setting: simultaneously generating both discrete sequence and continuous all-atom structural coordinates.
From structure prediction to real-world drug design Though recent works demonstrate promise for the ability of diffusion models to generate proteins, there still exist limitations of previous models that make them impractical for real-world applications, such as: All-atom generation: Many existing generative models only produce the backbone atoms. To produce the all-atom structure and place the sidechain atoms, we need to know the sequence.
This creates a multimodal generation problem that requires simultaneous generation of discrete and continuous modalities. Organism specificity: Proteins biologics intended for human use need to be humanized, to avoid being destroyed by the human immune system. Control specification: Drug discovery and putting it into the hands of patients is a complex process. How can we specify these complex constraints?
For example, even after the biology is tackled, you might decide that tablets are easier to transport than vials, adding a new constraint on soluability. Generating “useful” proteins Simply generating proteins is not as useful as controlling the generation to get useful proteins. What might an interface for this look like? For inspiration, let's consider how we'd control image generation via compositional textual prompts (example from Liu et al. , 2022). In PLAID, we mirror this interface for control specification.
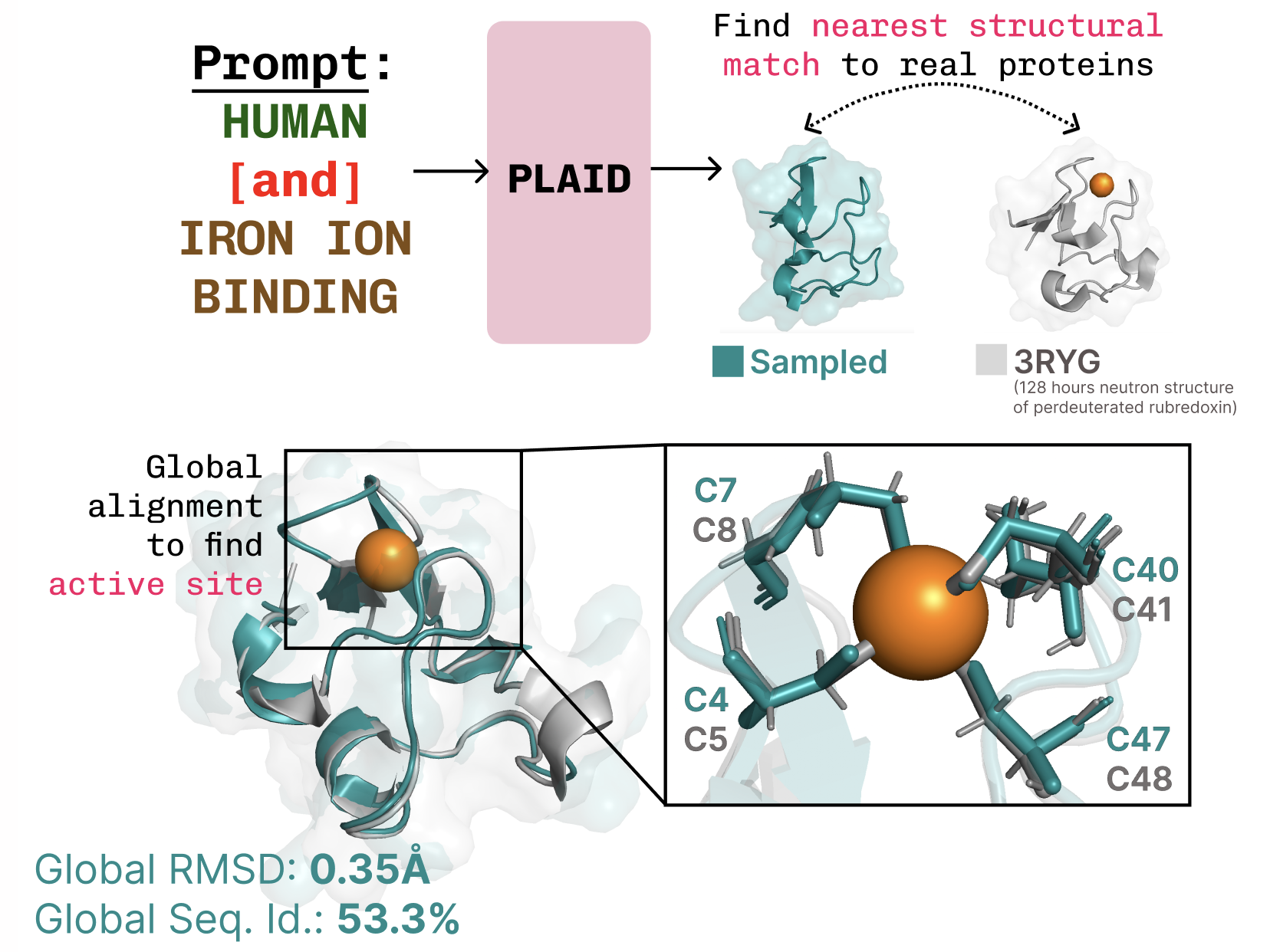
The ultimate goal is to control generation entirely via a textual interface, but here we consider compositional constraints for two axes as a proof-of-concept: function and organism: Learning the function-structure-sequence connection. PLAID learns the tetrahedral cysteine-Fe2+/Fe3+ coordination pattern often found in metalloproteins, while maintaining high sequence-level diversity.
Training using sequence-only training data Another important aspect of the PLAID model is that we only require sequences to train the generative model! Generative models learn the data distribution defined by its training data, and sequence databases are considerably larger than structural ones, since sequences are much cheaper to obtain than experimental structure. Learning from a larger and broader database.
The cost of obtaining protein sequences is much lower than experimentally characterizing structure, and sequence databases are 2-4 orders of magnitude larger than structural ones. How does it work? The reason that we’re able to train the generative model to generate structure by only using sequence data is by learning a diffusion model over the latent space of a protein folding model.
Then, during inference, after sampling from this latent space of valid proteins, we can take frozen weights from the protein folding model to decode structure. Here, we use ESMFold, a successor to the AlphaFold2 model which replaces a retrieval step with a protein language model. Our method. During training, only sequences are needed to obtain the embedding; during inference, we can decode sequence and structure from the sampled embedding. ❄️ denotes frozen weights.
In this way, we can use structural understanding information in the weights of pretrained protein folding models for the protein…
Want this in your inbox every morning?
Daily brief at your local 8am — bilingual EN/中文, free.